

A new global dataset containing 239 human-infectious RNA viruses shows how animal hosts, vector transmission, surveillance gaps, and viral characteristics shape the pathway from spillover to epidemic threat.

Research: A complete catalog of human infectious RNA viruses. Image credit: Andrzej Rostek / Shutterstock
Recent reviews published in magazines scientific data presents an updated global catalog of 239 ribonucleic acid (RNA) viruses known to infect humans, an increase of 25 from 2018, providing new insights into their emergence and spread.
Most viruses do not appear randomly but cluster within several families, are associated with non-human hosts, especially mammals, and are detected at varying rates over time as classification, reporting, surveillance, and sequencing technologies evolve.
Although human spillover is common worldwide, only a minority of species reach epidemic or endemic levels in humans, highlighting the critical bottleneck between exposure and epidemic spread.
RNA viruses continue to pose a growing threat to global health today, causing diseases such as measles, influenza, and AIDS, caused by HIV, and sparking new epidemics. Recent events related to Oroporchi virus and Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) highlight the epidemic potential of these viruses. However, the virus situation continues to change rapidly.
Almost every year, researchers identify new human-infecting species, revise classifications, and expand genomic and ecological data. As evidence about infection, host range, and spread accumulates, the need for an up-to-date catalog is critical to tracking what is known and predicting future risks.
Human RNA Virus Catalog Methodology
In this paper, researchers developed an updated and expanded dataset of RNA viruses known to infect humans that incorporates current knowledge through December 2024.
They conducted systematic literature searches every 1 to 3 years, based on previous catalogs from 2001 and 2018, using databases such as Web of Science, PubMed, Scopus, and Google Scholar, supplemented with sources such as the World Health Organization (WHO), Centers for Disease Control and Prevention (CDC), ProMed, and the National Center for Biotechnology Information (NCBI) genomic records.
This dataset includes only peer-reviewed primary reports that provide solid evidence that RNA viruses recognized by the International Committee on Taxonomy of Viruses (ICTV) infect humans under natural or real-world conditions, and does not include deliberate experimental inoculation or in vitro evidence.
The team resolved ambiguities through independent evaluation and consensus, and in some cases inferred properties that closely related viruses lacked. They compiled species-level data by integrating information across known subtypes and associated each virus with its first reported human case, genome sequence, and geographic origin.
Researchers used standardized criteria to record key characteristics such as transmissibility, host range, and route of transmission. They classified transmissibility into levels 2, 3, and 4, ranging from zoonotic diseases without human transmission to viruses that can cause epidemic or endemic transmission to humans.
Finally, the team mapped the date and location of discovery, allowing for temporal and spatial analysis of virus emergence. By integrating genomic, ecological, and epidemiological data into a single framework, the updated dataset provides a solid foundation for studying viral diversity, evolution, and public health risks.
The annual number of new and (currently) ICTV-recognized human infectious RNA virus species is shown in Figure 2a. Figure 2b shows the accumulation of species over time and the accumulation of genera and families that contain one or more human-infectious RNA virus species. The first human RNA virus, yellow fever virus, was described in 1901. The number of species increased slowly until the mid-1950s and somewhat faster thereafter. By the end of the 20th century, 178 species had been identified, and 61 more species had been added so far in the 21st century. Looking at the 10-year period, the 1960s introduced the most new species (42 species). This was followed by the 2000s (31 cases), but it declined again in the 2010s.
Global RNA virus discovery patterns
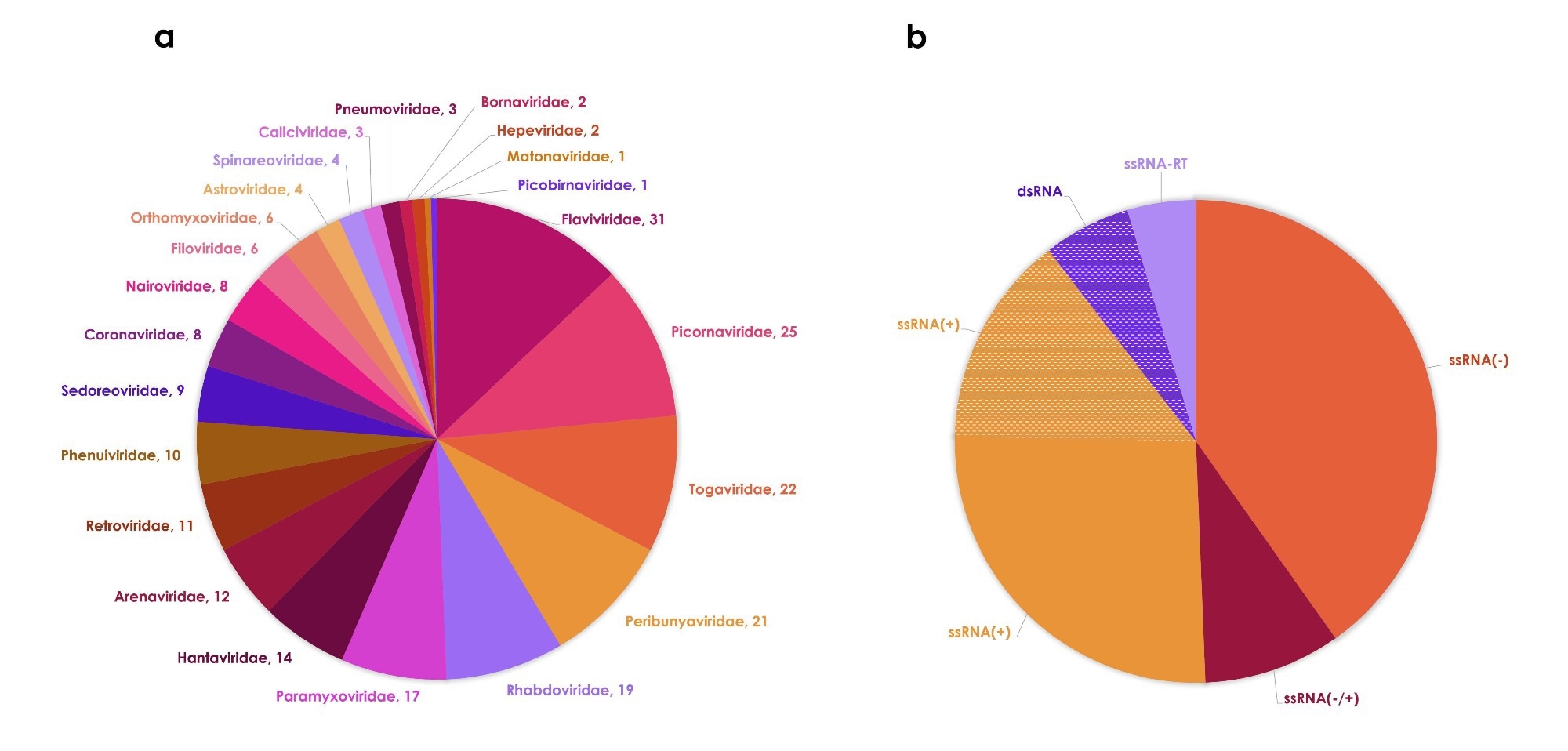
The updated dataset includes 239 RNA viruses classified by ICTV that are known to infect humans. Compared to 2018, this reflects the addition of 25 species identified through new discoveries and taxonomy updates.
Although these species span 61 genera and 23 families, diversity remains concentrated in a few families, and most viruses share common genomic features, particularly single-stranded RNA genomes.
The authors note that although discoveries have increased over time since the mid-20th century, formal analysis is needed to determine whether the overall discovery rate is increasing or decreasing.
After minimal growth in the early 20th century, identification rates rose sharply starting in the mid-1900s, with notable peaks in the 1960s and early 2000s. However, most newly identified species extend existing genera or families rather than introducing entirely new taxa.
Geographically, the first reported human cases have occurred on all inhabited continents, with clusters occurring in areas with stronger surveillance systems. This pattern highlights both the global nature of viral spillover and the impact of detection capacity on detection.
Possibility of spread, infection, and epidemic
Ecologically, the majority of viruses (62%) are strictly zoonotic (level 2), with no sustained human-to-human transmission. Only 60 species reach level 4. This means they are endemic to humans or can spread infectious diseases, and many of these species still maintain animal reservoirs.
Most viruses are associated with nonhuman mammalian hosts, reinforcing their central role in their emergence. Transmission routes are diverse, but vector-borne transmission is the most common, primarily through mosquitoes and ticks, followed by inhalation and direct contact.
In particular, transmission routes for a subset of viruses remain uncertain, reflecting persistent knowledge gaps. Taken together, these findings clearly highlight a situation defined by repeated and documented spillovers, escalation of discovery, and limited adaptation to sustained human infection.
RNA virus surveillance and risk prediction
These findings point to a more targeted and proactive approach to emerging viral threats. Rather than broadly searching for completely novel pathogens, future efforts could use this dataset to examine high-risk viral families, mammalian reservoirs, and areas with limited surveillance where undetected spillover is most likely to occur.
Expanding genome sequencing, metagenomics, and real-time monitoring will be critical to filling persistent knowledge gaps, especially regarding transmission routes and host range.
At the same time, this dataset provides a valuable basis for modeling trends in discovery and identifying traits associated with epidemic potential. As it continues to evolve, it will help refine risk predictions and guide early warning systems.
The ultimate challenge is not only to discover new viruses, but to understand which viruses are most likely to adapt, spread, and become the next global health threat.